química computacional.
La química computacional es todo aquel aspecto de la química el cual es explicado mediante el uso de un ordenador y un software.
De la química computacional se puede obtener la información molecular necesaria para describir un sistema, dado que es utilizada para conocer estados intermediarios de reacción, ángulos de enlace, propiedades electrónicas de la molécula, etc. En la actualidad existen programas específicos para realizar cálculos de química computacional, los cuales difieren en el método de cálculo, así como en la exactitud y recursos computacionales necesarios.
La química computacional nace con la necesidad de comprender aspectos importantes que no se pueden resolver de forma práctica, como lo es conocer estados intermediarios en reacciones que se efectúan rápidamente, conocer el comportamiento de un material a temperaturas muy altas que son muy difíciles de alcanzar en un laboratorio, o simplemente calcular propiedades en sistemas de alto costo.
El objetivo principal de la química computacional es predecir todo tipo de propiedades moleculares de sistemas químicos utilizando la fisicoquímica, la física molecular y la física cuántica, y emplea una gran variedad de técnicas teóricas en constante desarrollo.
metodos computacionales.
la quimica computacional abarca amplio uso de las matematicas complejas y estos amplios rangos matematicos pueden dividirse en dos grandes categorias.
la mecanica molecular.
Este método se basa en el modelado matemático de una molécula compuesta por átomos que se mantienen unidos por enlaces. Utiliza los parámetros de fuerza de tensión y flexión de enlace, lo cual permite interacciones entre los átomos no enlazados.
- la mecanica molecular es un metodo (esquema computacional) muy rapido para determinar geometria, energias moleculares, espectro vibracionales y entalpias de formacion de moleculas estables en su estado basal
- debido a su gran velocidad de calculo, es ampliamente usado para moleculas grandes.
- los enlaces entre particulas se compoprtan como osciladores armonicos.
- El núcleo y los electrones de un átomo están juntos, es decir, se trata como una partícula única.
- las particulas son tratadas como si fueran esferas

mecanica cuantica.
Los métodos de la mecánica cuántica describen las moléculas en términos de interacciones explícitas entre núcleos y electrones y se basan en los siguientes principios.
• Los núcleos y los electrones se distinguen unos de otros.
• Las interacciones electrón-electrón y electrón-núcleo son explícitas.
• Dichas interacciones están dirigidas por el movimiento y las cargas de los electrones.
• Las interacciones determinan la distribución espacial del núcleo, los electrones y sus energías.
• Los métodos de la mecánica cuántica resuelven mediante aproximaciones la ecuación de onda de Shrödinger.
ejemplo:
Funciones de onda del electrón en un átomo de hidrógeno a diferentes niveles de energía. La mecánica cuántica no puede predecir la ubicación exacta de una partícula en el espacio, solo la probabilidad de encontrarla en diferentes lugares. Las áreas más brillantes representan una mayor probabilidad de encontrar el electrón.

Comparación de los métodos de cálculo más utilizados en química computacional.

programas de la quimica compuacional.

SPARTAN:
Las funciones primarias son proporcionar información sobre estructuras, estabilidades relativas y otras propiedades de moléculas aisladas. Los cálculos de la mecánica molecular en moléculas complejas son comunes en la comunidad química. Los cálculos químicos cuánticos, incluidos los cálculos orbitales moleculares del método Hartree-Fock , pero especialmente los cálculos que incluyen la correlación electrónica , requieren más tiempo en comparación.

TAREAS REALIZADAS:
Energía
|
para una geometría dada, proporciona energía y propiedades asociadas de una molécula o sistema. Si se emplean modelos químicos cuánticos, se calcula la función de onda .
|
Geometría molecular de equilibrio
|
ubica el mínimo local más cercano y proporciona energía y propiedades asociadas.
|
Geometría del estado de transición
|
ubica el punto de asiento de primer orden más cercano (un máximo en una sola dimensión y mínimos en todos los demás) y proporciona energía y propiedades asociadas.
|
Conformador de equilibrio
|
localiza la conformación de energía más baja. A menudo se realiza antes de calcular la estructura utilizando un modelo químico cuántico.
|
Obtiene una selección de conformadores de baja energía. Comúnmente utilizado para identificar las formas que probablemente adopte una molécula específica y para determinar una distribución de Boltzmann para calcular las propiedades moleculares promedio.
| |
Biblioteca de conformadores
|
ubica al conformador de menor energía y lo asocia con un conjunto de conformadores que abarcan todas las formas accesibles a la molécula sin importar la energía. Se utiliza para construir bibliotecas para el análisis de similitud.
|
pasa una molécula o sistema a través de un conjunto de coordenadas definidas por el usuario, proporcionando geometrías de equilibrio para cada paso (sujeto a restricciones especificadas por el usuario).
| |
Análisis de similitud
|
cuantifica la similitud de las moléculas (y, opcionalmente, sus conformadores) en función de la estructura o la función química ( aceptadores de enlaces de hidrógeno -donantes, ionizables positivo-negativo , hidrófobos , aromáticos ). Cuantifica la semejanza de una molécula (y opcionalmente sus conformadores) a un farmacóforo .
|
Modelos gráficos
superficies:
Un conjunto de orbitales moleculares y la energía de cada uno de ellos. Para construir la función de onda total situaremos los electrones en los orbitales moleculares siguiendo el orden de sus energías. Las energías de los orbitales moleculares nos permiten, por lo tanto, conocer el orden de "llenado" de los orbitales y la configuración electrónica.
|  | |
la densidad, ρ ( r ), es una función de las coordenadas r , definida de tal manera que ρ ( r ) d r es el número de electrones dentro de un pequeño volumen d r . Esto es lo que se mide en un experimento de difracción de rayos X. La densidad se puede representar en términos de una isosuperficie (superficie de isodensidad) con el tamaño y la forma de la superficie dados por el valor (o porcentaje de envolvente) de la densidad electrónica.
|  | |
la densidad, ρ spin ( r ), se define como la diferencia en la densidad de electrones formada por electrones de α spin, ρα ( r ), y la densidad de electrones formada por electrones de β spin, ρβ ( r ). Para las moléculas de capa cerrada (en las que todos los electrones están emparejados), la densidad de espín es cero en todas partes. Para las moléculas de cubierta abierta (en la que uno o más electrones están desapareados), la densidad de espín indica la distribución de los electrones no apareados. La densidad del espín es un indicador de reactividad de los radicales.
| ||
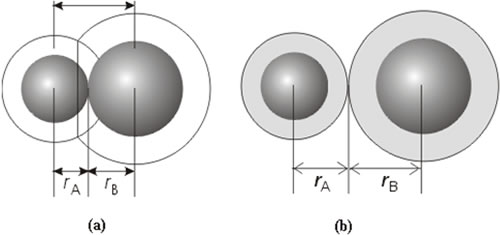
si dos átomos de gases nobles se fuerzan a estar juntos sin energia cinetica que los separe,van a permanecer unidos. Siendo las fuerzas electrostáticas débiles entre dipolos las que lo mantienen
|  | |
 | ||
el potencial, ε p , se define como la energía de interacción de una carga puntual positiva ubicada en p con los núcleos y electrones de una molécula. Una superficie para la cual el potencial electrostático es negativo (una superficie de potencial negativo) delinea las regiones de una molécula que están sujetas a un ataque electrofílico.
|  |
Superficies compuestas (mapas) :
Mapa de potencial electrostático (indicador electrofílico)
|
el mapa de propiedades más comúnmente empleado es el mapa de potencial electrostático. Esto da el potencial en ubicaciones en una superficie particular, más comúnmente una superficie de densidad electrónica que corresponde al tamaño molecular global.
|  |
Mapa de potencial de ionización local
|
se define como la suma sobre las densidades de electrones orbitales, ρi ( r ) veces las energías orbitales absolutas, ∈i, y se divide por la densidad electrónica total, ρ ( r ). El potencial de ionización local refleja la relativa facilidad de eliminación de electrones ("ionización") en cualquier ubicación alrededor de una molécula. Por ejemplo, una superficie de potencial de ionización local "bajo" para el tetrafluoruro de azufre desmarca las áreas que se ionizan más fácilmente.
| |
Mapa LUMO (indicador nucleófilo)
|
los mapas de orbitales moleculares también pueden conducir a indicadores gráficos. Por ejemplo, el mapa de LUMO , en el que el (valor absoluto) del orbital molecular más desocupado (el LUMO) se asigna a una superficie de tamaño (de nuevo, más comúnmente la densidad electrónica ), proporciona una indicación de la reactividad nucleofílica.
|  |
No hay comentarios:
Publicar un comentario